
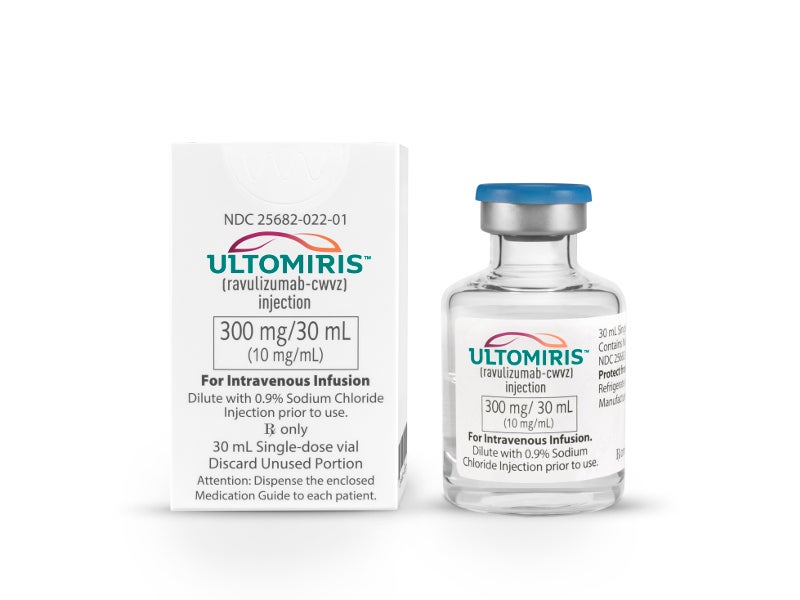
Ultomiris™ (ravulizumab-cwvz) is a complement inhibitor drug approved for the treatment of paroxysmal nocturnal haemoglobinuria (PNH) in adults.
In June 2018, the company submitted a marketing authorisation application to the European Medicines Agency (EMA), which is now under review. The drug is also under review in Japan.
Recommended Buyers Guides
Alexion Pharmaceuticals also submitted a biologics license application (BLA) for the drug to the US Food and Drug Administration (FDA) in June 2018. The FDA accepted the BLA under priority review status in August 2018 and approved it in December 2018.
Ravulizumab has secured orphan drug designation in the US and European Union for PNH treatment.
Alexion Pharmaceuticals is evaluating the drug for the treatment of atypical hemolytic uremic syndrome (aHUS) and plans to initiate a clinical study for the treatment of generalised myasthenia gravis (MG).
Paroxysmal nocturnal haemoglobinuria causes and symptoms
PNH is a progressive blood disorder that causes the premature destruction of red blood cells due to uncontrolled activation of an immune system component called the complement system.
PNH patients may experience a wide range of symptoms, including fatigue, dysphagia, dyspnoea, pain in the abdomen, erectile dysfunction, anaemia and dark-coloured urine. The disease may also cause thrombosis, which leads to the dysfunction of vital organs.
An estimated 20% to 35% of patients die within five to ten years of disease diagnosis.
Ultomiris’ mechanism of action
Ultomiris is a humanised monoclonal antibody (mAb) produced in Chinese hamster ovary cells. It is a terminal complement inhibitor that specifically binds to the C5 protein of the terminal complement cascade and inhibits its breakdown. The drug prevents the destruction of terminal complement-mediated red blood cells in patients with PNH.
Ultomiris is available as a sterile preservative-free solution in single-dose vials for intravenous administration. Each vial contains 300mg/30ml (10mg/ml) of ravulizumab-cwvz concentration at pH 7.
Clinical trials on Ultomiris
The FDA’s approval of Ultomiris was based on the positive results of two 26-week, open-label, multi-centre, randomised, active-controlled, non-inferiority phase three clinical studies named PNH Study 301 and PNH Study 302. 441 patients were enrolled in the two studies.
The PNH Study 301 enrolled 246 previously untreated PNH patients with complement inhibitor. The patients were randomised in a 1:1 ratio to receive either Ultomiris or eculizumab (Soliris®). Transfusion avoidance and haemolysis were determined to evaluate the efficacy of the drug.
The transfusion avoidance rate in patients treated with Ultomiris™ was 73.6% compared with 66.1% in those treated with Soliris®. Just 4% of the patients treated with Ultomiris™ showed breakthrough haemolysis, compared with 10.7% of patients treated with Soliris.
An estimated 68% of patients treated with Ultomiris demonstrated stabilised haemoglobin, compared with 64.5% of patients treated with Soliris.
The PNH Study 302 trial enrolled 195 PNH patients previously treated with Soliris. The patients were randomised in a 1:1 ratio to receive either Ultomiris or Soliris. The primary endpoint of the study was haemolysis, while other factors such as transfusion avoidance rate and haemoglobin stabilisation were also determined to evaluate the efficacy of the drug.
The patients receiving Ultomiris did not show any breakthrough haemolysis compared to 5.1% Soliris-treated patients.
The transfusion avoidance rate in Ultomiris-treated patients was 87.6% compared with 82.7% in Soliris-treated patients. An estimated 76.3% Ultomiris-treated patients showed stabilised haemoglobin in comparison with 75.5% of Soliris-treated patients.
Upper respiratory infection and headaches were the most common adverse events noticed in patients during the trial.